ClimAHealth
This project is maintained by dtortosa
Summary of the project
After migrating out of Africa, human populations were exposed to a wide range of environmental conditions and, particularly, temperatures. Over millennia, exposure to varying temperatures could have modified the response to external temperature in these populations. In other words, this exposure could act as a selective pressure, shaping the metabolism of human populations and leading them to adapt to different temperature regimes. This process leaves detectable signals in the genome, known as signatures of positive selection or adaptation. We can identify these signals using various statistical and machine learning methods.
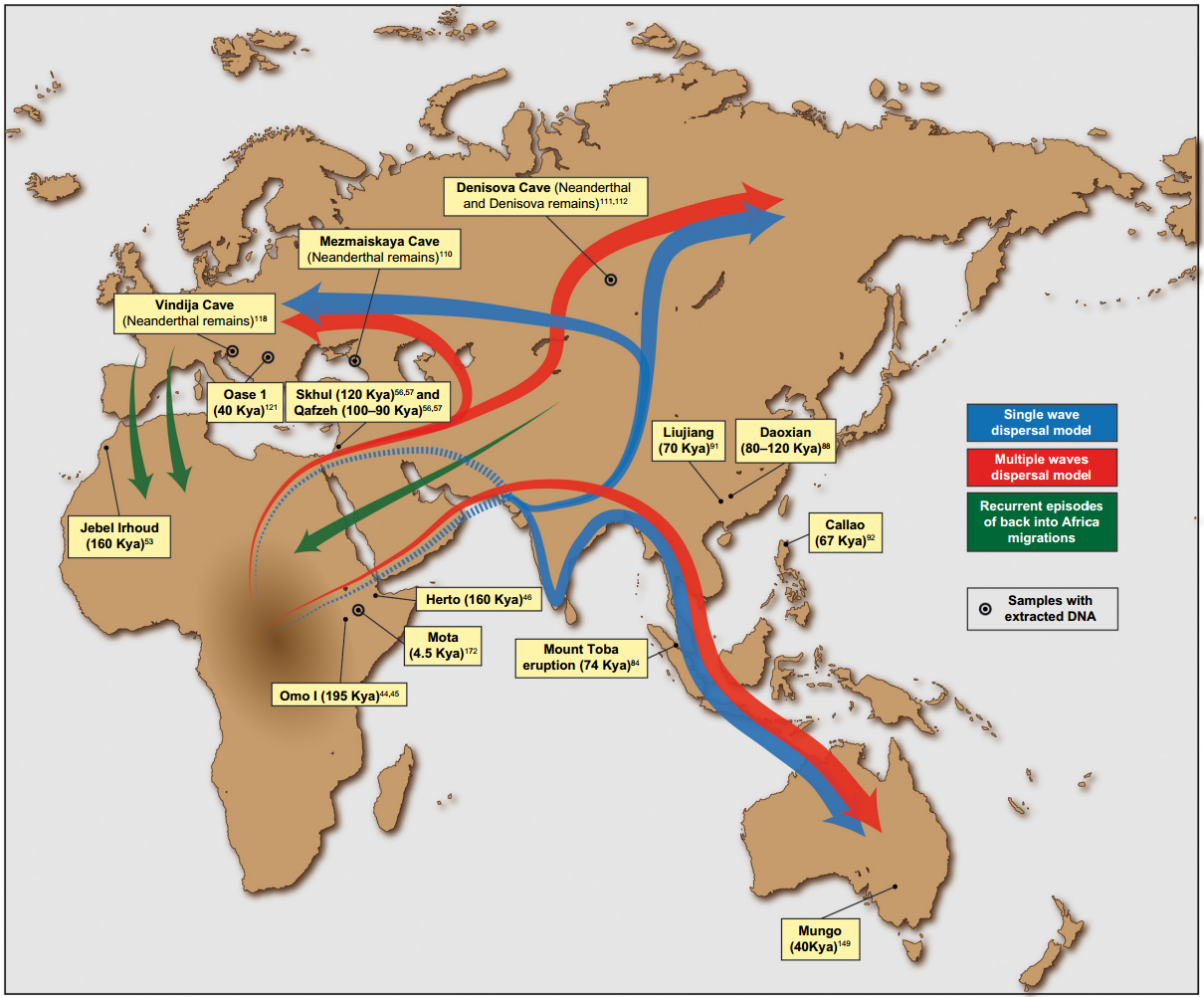 Possible migration waves out of Africa along with locations of major ancient human remains and archeological sites. Saioa López, Lucy van Dorp and Garrett Hellenthal - López, S., van Dorp, L., & Hellenthal, G. (2015). Human Dispersal Out of Africa: A Lasting Debate. Evolutionary Bioinformatics Online, 11(Suppl 2), 57–68. Link doi, link ncbi, CC BY 3.0.
Possible migration waves out of Africa along with locations of major ancient human remains and archeological sites. Saioa López, Lucy van Dorp and Garrett Hellenthal - López, S., van Dorp, L., & Hellenthal, G. (2015). Human Dispersal Out of Africa: A Lasting Debate. Evolutionary Bioinformatics Online, 11(Suppl 2), 57–68. Link doi, link ncbi, CC BY 3.0.
Considering that the mechanisms for coping with temperature changes involve energy production and the use of different energy substrates like glucose or fatty acids, they are closely related to energy balance and cardiometabolic traits. Consequently, past adaptive events that altered the ability of human populations to expend energy in heat production might contribute to differential risk of metabolic disorders between current populations.
ClimAHealth was dedicated to the search of signals of adaptation caused by climate across human populations. We combined statistics sensitive to adaptation with a diverse range of machine learning approaches. This allowed us to test whether signals of adaptation are more frequent in genes implicated in energy metabolism and hence relevant for coping with temperature changes. An excess of adaptation signals within these genes would suggest the existence of past adaptation to temperature conditions. Furthermore, this would support the functional relevance of genetic variants within these energy metabolism genes, i.e., their impact on human traits. For a genetic variant to undergo positive selection, it should impact a trait that influences an individual’s ability to cope with external factors and contribute to reproductive success. Given the connection of these genes with energy balance, ClimAHealth will also assess whether they tend to associate with obesity-related traits.
Work performed and main results achieved so far
ClimAHealth has focused on the detection of recent signatures of adaptation (left in the last 30,000 years). We calculated the probability of adaptation across human genes using a well-known statistic for recent adaptation (the integrated haplotype score; iHS). We also quantified genomic characteristics around these genes that should associate with adaptation if the latter was common enough in the first place. In other words, if adaptation was relatively frequent during recent human evolution, signals of adaptation should occur more frequently in genomic regions that are more likely to influence human phenotypes. In contrast, if signals of adaptation are just false positives, then they should be randomly distributed across the human genome.
We have developed and applied a modeling framework able to predict the probability of adaptation (iHS) using multiple predictors as input. In other words, we have been able to assess the influence of multiple genomic factors on adaptation at the same time. This framework considered a wide range of algorithms from simple linear models to deep neural networks. It applied a rigorous training/evaluation scheme in order to, first, select the best combination of parameters within a given algorithm category and then make a final selection between the best candidates across all categories. The models were evaluated and tested against data not previously seen in order to obtain a rigorous estimate of model performance. Given the high computational burden of this model exploration, we took advantage of initial results of ClimAHealth showing that adaptation signals were more visible in the African Yoruba population compare to non-African populations. Therefore, Yoruba was a good population to battle-test our framework and find the best algorithm and architecture to detect true iHS selection signals. This was a deep neural network with 5 layers and 1000 nodes (among other parameters) that then was used to model iHS in completely different and new populations (25 populations spread across 4 continents). This approach achieved a high predictive power explaining ~80% of the variability of iHS when predicting on unseen data. Notably, we identified multiple genomic factors associated with adaptation across all populations. This suggests that signals of recent adaptation were not randomly distributed across the genome, reducing the likelihood of them being false positives. In other words, according to these results, adaptation should have been relatively frequent in human populations in the past 30 Kya. Importantly, we included as predictor in the models the distance to genes related to thermoregulation in general, and thermogenesis in skeletal muscle and brown adipose tissue in particular. We found a higher probability of adaptation around SMT and BAT genes compared to the rest of the genome across most of the studied populations. Given the link between thermogenesis and environmental temperature, these adaptation signals provide evidence for climate adaptation in humans. This also suggests that genetic variants in these thermogenic genes are functionally relevant, as signals of adaptation should occur in genomic regions impacting human phenotypes.
In the last step of this project, we performed a genome-wide association study using as input genetic variants coming from individuals exposed to a training regime where cardio-respiratory performance and body mass were measured before and after. We applied multiple quality control procedures required to reduce confounding factors like population structure or sample relatedness along with an imputation step in the TOPMed imputation server resulting in 3M variants for 1K samples. The processed data was then used to calculate the association between all genetic and the traits under study (weight change after the training regime). This was then used to calculate the average level of association for all genetic variants within thermogenic genes and compare it with randomly selected variants across the genome, which acted as the random expectation. We found that thremogenic genes had a higher association with weight change than expected by chance. Therefore, we have defined a set of genes related to thermoregulation that exhibited an accumulation of adaptation signals and, at the same time, significantly correlate with body mass. This suggests the existence of past events of climate-related adaptation that shaped the physiology of human populations and, in turn, this could influence the variability of health-related traits like body mass.
The main scripts used to perform these analyses can be found in the following links:
- Defintion and validation of the BAT connectome (link).
- Mixture Density Regression to model iHS (link).
- Modeling framework to compare a battery of algorithms and select the best to model iHS across 26 human populations (link).
- Association between PTPN1 genetic variants and obesity-related traits (link).
Progress beyond the state of the art
ClimAHealth is advancing the analysis of adaptation signals beyond the state of the art. Previous work in this field has relied on classical correlation and regression approaches, which do not align well with the distribution of adaptation statistics. Leveraging a novel modeling framework, we analyze complex adaptation statistics, considering the impact of multiple factors on the probability of adaptation.
The heterogeneity of the human genome has greatly complicated the search for recent adaptation signals, resulting in a controversial topic. However, our innovative modeling approaches have provided robust evidence indicating relatively frequent adaptation in recent evolutionary times. As a result, ClimAHealth is contributing to answering critical questions in human evolutionary biology. It is also opening new avenues of research by applying novel modeling techniques to model positive selection.
Last, but not least, we defined a list of genes related to brown adipose tissue that was initially validated, confirming its cohesion and relationship with BAT. Many genes included in this list were already known to be directly implicated in BAT, but not all of them, being the latter potential novel candidates. The presence of an accumulation of adaptive signals within this group of genes further support the functional relevance of these genes making it a promising list of novel candidates to improve our knowledge about BAT. This has broader implications given the influence of BAT in glucose and plasma lipids, being these candidates potentially relevant for the genetic architecture of health-related traits.
Conclusions
In this project, we have developed and applied novel machine learning approaches to model signals of adaptation in the human genome. In general, we found an accumulation of adaptation signals in genes related to thermoregulation and, in particular, in genes related to thermogenesis in skeletal muscle and brown adipose tissues (SMT and BAT). This accumulation was present in multiple human populations across 4 continents, suggesting the existence of multiple events of adaptation to climate in the last 30,000 years. Additionally, thermogenic genes showed a higher than expected association with body mass in an independent genome-wide association study (GWAS). In sum, these findings provide evidence about widespread events of adaptation to climate across human populations along with potential implications of these events for cardiometabolic health.
Funding
This project is funded by the European Union’s Horizon 2020 research and innovation programme (Marie Skłodowska-Curie Actions; grant agreement number 101030971). This projects only reflects the author’s view and the Agency and the Commission are not responsible for any use that may be made of the information it contains.
![]()